
Определение остатков малахитового зеленого в пищевых продуктах животного происхождения, выращенных в водной среде, методом ВЭЖХ-МС/МС
- Главная страница
- Пресс-центр
- Статьи
- Определение остатков малахитового зеленого в пищевых продуктах животного происхождения, выращенных в водной среде, методом ВЭЖХ-МС/МС
1. Введение
Малахитовый зеленый, и его основной метаболит лейкомалахитовый зеленый и другие трифенилметановые соединения широко используются в качестве фунгицидов и противопаразитарных препаратах в процессе разведения аквакультуры. Эти вещества обладают мутагенными, тератогенными и канцерогенными свойствами. Многие страны запретили их использование в ветеринарии. В 2011 году был принят Технический регламент Таможенного союза «О безопасности пищевой продукции» № ТР ТС 021/2011 на пищевую продукцию, который запрещает остаточное содержание трифенилметановых красителей в товарной рыбе. Однако, из-за недостатка эффективных аналогов и интереса производств, нелегальное использование малахитового зеленого продолжается. Быстрые, чувствительные и точные методы обнаружения являются необходимым инструментом в усилении контроля над запрещенным веществом в ветеринарии. На сегодняшний день количественное содержание малахитового зеленого определяется согласно ГОСТ Р 57025-2016 «Рыба, нерыбные объекты и продукция из них. Иммуноферментный метод определения остаточного содержания трифенилметановых красителей». В тоже время остатки малахитового зеленого и его основного метаболита в продуктах водного происхождения могут быть определены с помощью эффективного и высокочувствительного метода ультра- эффективной жидкостной хроматографии с тройным квадрупольным тандемным масс-спектрометром технологии EXPEC.
2. Эксперимент
2.1 Стандартные образцы, реагенты и оборудование
Стандарты: стандарт 2-х веществ – малахитовый зеленый и его внутренние стандарты приобретены у Shanghai Anpel и хранились в холодильнике при -20°C. Реагенты: метанол ацетонитрил хроматографически чистые, муравьиная кислота хроматографически чистая, процентное содержание веществ в растворе от 98%.
Оборудование: УВЭЖХ 510 ультра- высокоэффективный жидкостной хроматограф (специально оснащен инфузионным бинарным насосом ультра-высокого давления, автосамплером ультра-высокого давления (с функцией охлаждения) и термостатом для колонки), EXPEC 5210 трехквадрупольный масс-спектрометр.
2.2 Условия УВЭЖХ и масс-спектрометрического детектирования
|
УВЭЖХ условия |
Подвижная фаза | 0.1% водный раствор муравьиной кислоты (A) и ацетонитрил (B), градиентное элюирование | ||
| Скорость потока | 0,3 мл/мин | |||
| Хроматографическая колонка | Waters BEH C18 (2.1*100мм, 1.7мкм) | |||
| Объем инжектирования | Режим: частичного заполнения петли, 3 мкм | |||
| Температура колонки | 40°C | |||
| Время анализа | 6 мин | |||
| Градиент | Время (мин) | A(%) | B(%) | |
| 0 | 90 | 10 | ||
| 1.5 | 90 | 10 | ||
| 2.5 | 5 | 95 | ||
| 3.5 | 5 | 95 | ||
| 3.6 | 90 | 10 | ||
| 6 | 90 | 10 | ||
| Условия МС | Режим работы | Регистрация положительных ионов | ||
| Поток распыляющего газа | 1,4 л/мин | |||
| Скорость потока газа десольватации | 4 л/мин | |||
| Обратный поток газа 2 л/мин | 2 л/мин | |||
| Температура газа десольватации | 500°C | |||
| Встречный поток газа | 0.45 мл/мин (5.8e-3Torr) | |||
| Напряжение на капилляре | 4.8 кВ | |||
Режим мониторинга – ММР (мониторинг множественных реакций). Параметры пар ионов, времени сканирования (dwell time), потенциал декластеризации (cone voltage) и энергия соударения (collision energy) для мониторинга приведены в таблице ниже.
| № | Вещество | Ионные пары | Время сканирования (с) | Потенциал декластеризации (В) | Энергия соударения (eV) | |
| 1 | Малахитовый зеленый | 329.25 | 313.25 | 0,06 | 50 | 38 |
| 208.15 | 50 | |||||
| 2 | Малахитовый зеленый-d5 | 334.15 |
318.25 |
0,06 | 50 |
41 |
| 3 | Лейкомалахитовый зеленый | 331.15 | 316.25 | 0,06 | 50 | 20 |
| 239.15 | 38 | |||||
| 4 | Лейкомалахитовый зеленый-d5 | 337.25 |
322.25 |
0,06 | 50 |
22 |
2.3 Подготовка образца
Первостепенно проводили экстракцию образца. Отбирали 5,00 г образца в 50 мл центрифужную пробирку, добавляли 1,5 мл 20% раствора гидроксиламина, 4 ил 1,0 М раствора пара- толуолсульфоновой кислоты и 5,0 мл буферного раствора ацетата. Содержимое пробирки гомогенизировали со скоростью 10000 об/мин в течении 30 секунд. Затем добавляли 10 мл ацетонитрила и интенсивно встряхивали в течение 30 секунд. После добавляли 5 г кислотного оксида алюминия и снова встряхивали в течение 30 секунд. Далее центрифугировали при 3000 об/мин в течение 10 минут. Супернатант переносили в 100 мл центрифужную пробирку, содержащую 10 мл воды и 2 мл диэтиленгликоля. Затем добавляли 10 мл ацетонитрила в 50 мл центрифужную пробирку и повторяли процесс экстракции, объединяя слои ацетонитрила.
Затем проводили очистку экстракта. К полученному экстракту добавляли 15 мл дихлорметана и встряхивали в течение 10 секунд, затем центрифугировали при 3000 об/мин в течение 10 минут. После слой дихлорметана переносили в 100 мл колбу, повторяли операцию с добавлением 5 мл ацетонитрила и 10 мл дихлорметана. Слои дихлорметана объединяли в 100 мл колбе. Полученный раствор испаряли при 45 °C до остатка объема в 1 мл, который растворили в 2,5 мл ацетонитрила.
Колонку с кислотным оксидом алюминия (перед применением активируется 5 мл ацетонитрилом) помещали на установку для твердофазной экстракции. Переносили раствор в колонку, затем дважды промывали колбу ацетонитрилом по 2,5 мл. Экстракт пропускали через колонну, контролируя скорость потока не более 0,6 мл/мин, и собирали весь выходящий раствор. Затем очищенный экстракт испарили при температуре 45 °C до почти сухого состояния, остаток растворяли в 0,5 мл ацетонитрила и пропускали через фильтровальную мембрану с диаметром пор 0,45 мкм. Фильтрат использовали для анализа.
3. Результаты
3.1 Линейность и предел обнаружения
Разбавление стандарта проводилось ацетонитрилом для достижения серии стандартных растворов (0,5 мкг/мл, 1 мкг/мл, 5 мг/мл, 10 мг/мл и 50 мкг/мл) малахитового зеленого. Концентрация внутреннего стандарта малахитового зеленого-d5 и лейкомалахитового зеленого-d5 составляла 6 мкг/мл.
Стандарты анализировали согласно описанному методу. Для построения калибровочной кривой по оси ординат (Y) расположено отношение площади хроматографического пика выбранного характеристического иона для каждого исследуемого компонента к площади пика соответствующего внутреннего стандарта.
По оси абсцисс (Х) отмечена массовая концентрация раствора определяемого стандарта и для нормализации кривой был применен метод весовых коэффициентов по формуле 1/х. Калибровочная кривая, построенная по методу внутреннего стандарта приведена на рис. 1 и 2. Хроматограмма характеристического иона целевых веществ в нижней точке калибровочной кривой показана на рис. 3.

Рис. 1 Калибровочная кривая малахитового зеленого

Рис. 2 Калибровочная кривая лейкомалахитового зеленого
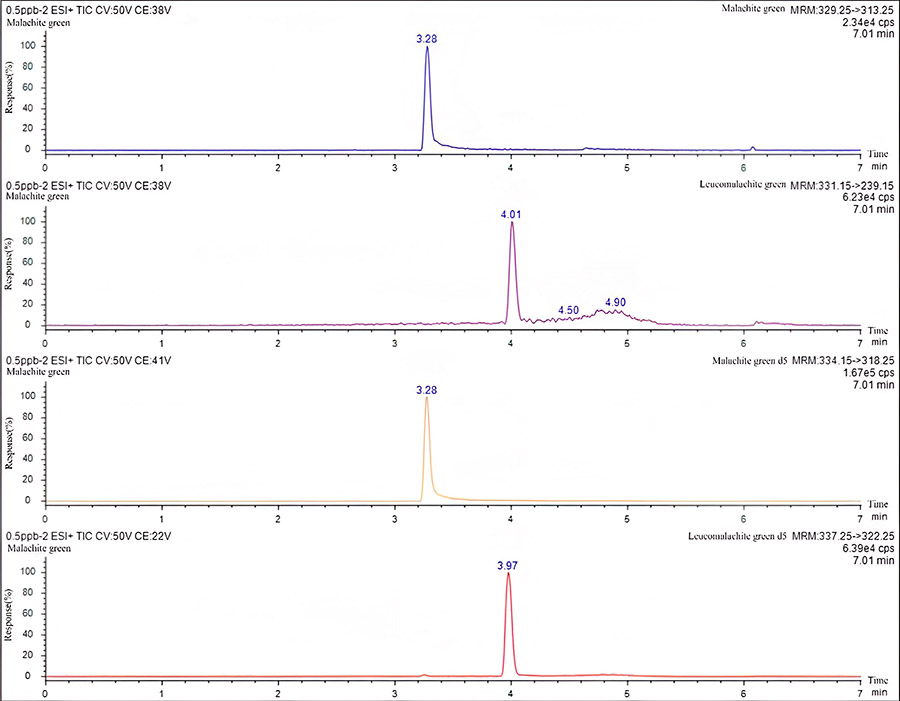
Рис. 3 Хроматограмма в точке предела количественного определения малахитового зеленого (1 мкг/мл,в котором внутренний стандарт малахитового зеленого-d5 и лейкомалахитового зеленого-d5 в концентрации 6 мкг/мл).
3.2 Прецизионность
Для подтверждения сходимости результатов анализа (время удерживания вещества и площадь пика) были параллельно проанализированы растворы стандартных веществ с концентрацией 0,5 мкг/мл и 5 мкг/мл в 6-ти повторностях для каждой концентрации. Полученные результаты приведены в таблицах ниже.
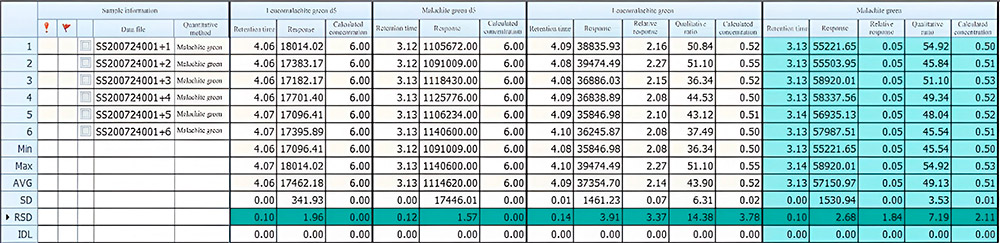
Таблица 2. Сходимость результатов анализа при концентрации 0,5 мг/мл: значение относительного стандартного отклонения (RSD) времени удерживания и отклика составило 0.10%-0.14% и 1.84%-3.91% соответственно.
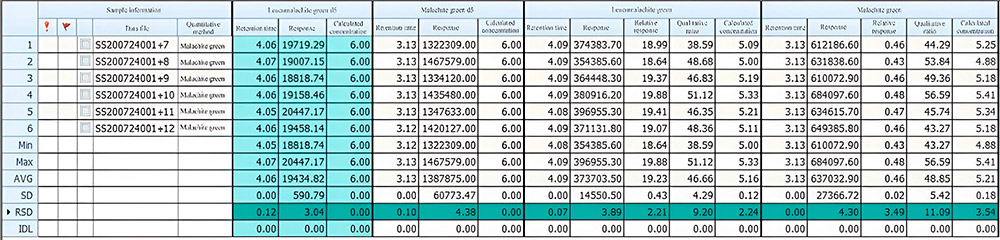
Таблица 3. Сходимость результатов анализа при концентрации 5 мг/мл: значение относительного стандартного отклонения (RSD) времени удерживания и отклика составило 0.07%-0.12% и 3.04%-4.30%соответственно.
По полученным данным стоит отметить хорошую прецизионность результатов анализа по параметрам времени удерживания и площади пика исследуемых веществ.
4. Вывод
После экстракции образцы пищевых продуктов животного происхождения, выращенных в водной среде отчищают с применением колонки, содержащей нейтральный оксид алюминия, а также используют другие процессы предварительной обработки для последующего определения экстрактов на УВЭЖХ-МС-МС. В работе приведены результаты по линейности, повторяемости и чувствительности метода. Полученные данные отражают линейность построенной калибровочной кривой для малахитового зеленого в диапазоне обнаружения, с коэффициентом корреляции более 0,99; отклонение сходимости результатов находится в пределах 4,3%. Система EXPEC5210 может быть использована для эффективного количественного определения остаточного содержания малахитового зеленого в пищевых продуктах животного происхождения, выращенных в водной среде.
и приглашать на предстоящие выставки и семинары